
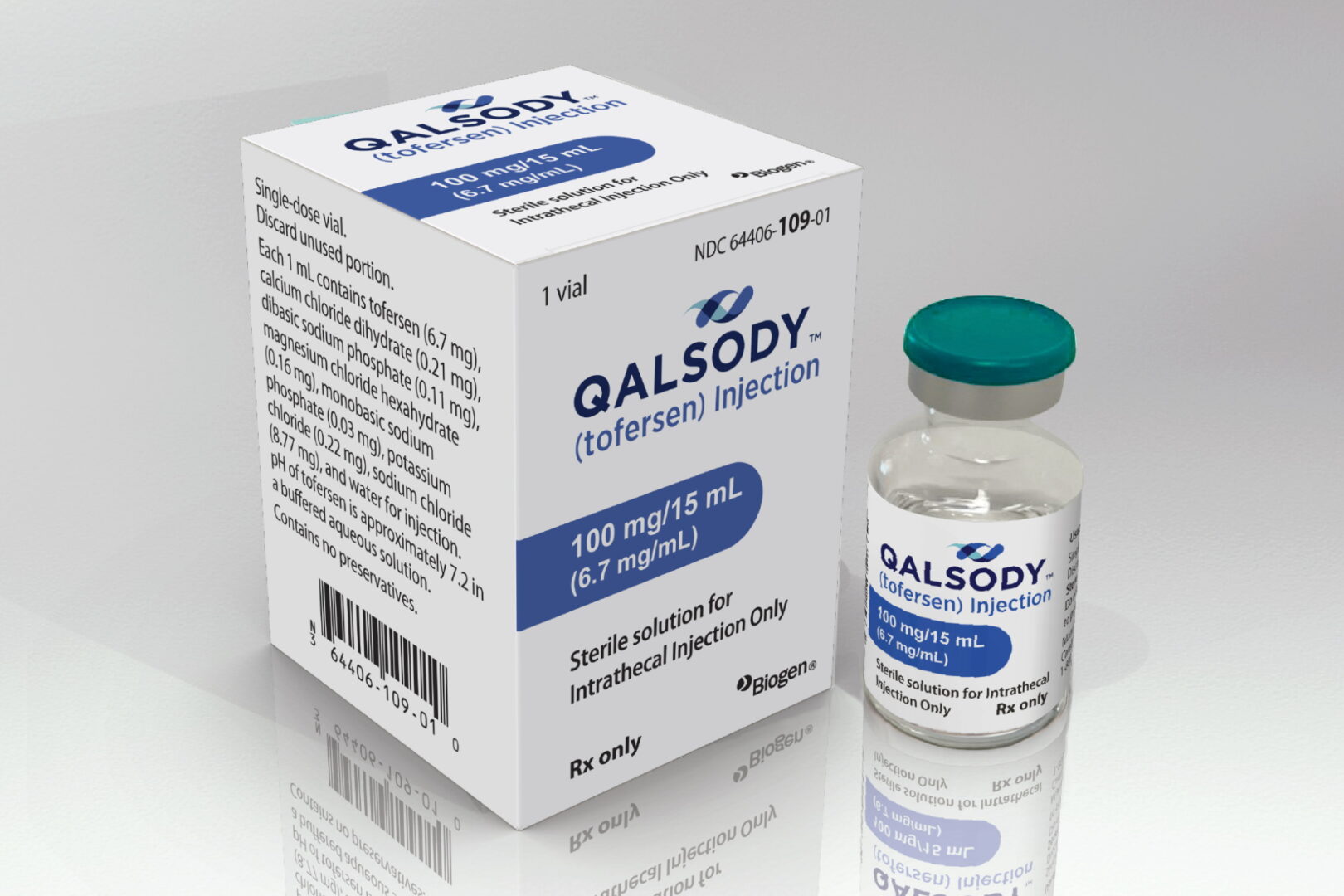
Le 1er traitement ciblant la cause génétique de la maladie de Charcot ou sclérose latérale amyotrophique (SLA), QALSODY™ (tofersen), vient d’être approuvé dans le cadre d’une procédure accélérée par la FDA aux États-Unis. Révolutionnaire.
Biogen a annoncé le 25 avril 2023 que la Food & Drug Administration (FDA) a autorisé QALSODY™ (tofersen) pour traiter la sclérose latérale amyotrophique (SLA), aussi appelée maladie de Charcot, chez les adultes avec une mutation du gène SOD1. C’est la plus fréquente des maladies neurogérénatives L’autorisation a été accordée rapidement car QALSODY™ a montré une réduction d’une protéine, appelée neurofilament, qui est un indicateur de dégradation des neurones. Une étude de phase 3, nommée ATLAS, est en cours pour confirmer les bienfaits du médicament et pourrait permettre une autorisation permanente.
Qu’est-ce que la maladie de Charcot ?
La maladie de Charcot est une affection neurologique progressive qui affecte les neurones moteurs, responsables de la transmission des signaux du cerveau aux muscles. Elle a été décrite pour la première fois par le neurologue français Jean-Martin Charcot en 1869. Charcot a observé et étudié les symptômes de cette maladie chez plusieurs patients, et il a publié ses observations dans un article intitulé « De la sclérose latérale amyotrophique » (SLA). Cette maladie est aujourd’hui connue sous le nom de maladie de Charcot en l’honneur de son découvreur. Elle touche entre 7000 et 8000 personnes en France, soit 1 personne sur 25 000 en se déclenchant entre 50 et 70 ans. Et tous les jours, 4 personnes meurent, tous les jours, en France de cette maladie neurodégénérative.
Lorsque les neurones sont endommagés, les muscles perdent progressivement leur capacité à fonctionner, entraînant une paralysie et une perte de mobilité. Cette maladie neurodégénérative rare touche les nerfs moteurs.
Les personnalités touchées (ou ayant eu) par la maladie de Charcot :
- Stephen Hawking,
- Jean-Yves Lafesse,
- Frank Alamo,
- Marc-Antoine Le bret,
- Charles Bietry,
- Doully,
- Thierry Monfray,
- Pone (Fonky Family)
Pourquoi la maladie de Charcot est-elle une maladie génétique ?
Dans la plupart des cas, la sclérose latérale amyotrophique (SLA) est sporadique, ce qui signifie qu’elle survient sans antécédents familiaux. Cependant, environ 5 à 10% des cas de SLA sont héréditaires, ce qui signifie qu’ils sont causés par des mutations dans des gènes spécifiques transmis de génération en génération. Elle est considérée comme une maladie génétique en raison de son association avec des mutations dans plusieurs gènes importants pour la santé des neurones moteurs.
Un grand nombre de gènes jouent un rôle dans l’apparition de la sclérose latérale amyotrophique (SLA). Le dépistage génétique peut permettre d’identifier si la maladie est causée par une mutation génétique, y compris chez les individus sans antécédents familiaux avérés. La SLA-SOD1 affecte près de 2 % des patients atteints de SLA, ce qui représente environ 330 personnes aux États-Unis et moins de mille en Europe. Il est estimé que plus de 15 % des personnes atteintes de la SLA présentent une forme génétique de la maladie. Toutefois, certaines de ces personnes ne présentent pas d’antécédents familiaux connus.
Les mutations les plus courantes associées à la SLA héréditaire sont celles dans les gènes SOD1, C9ORF72, TARDBP, FUS et UBQLN2. Ces gènes fournissent des instructions pour la production de protéines qui sont importantes pour la santé des neurones moteurs, qui sont les cellules nerveuses qui contrôlent les mouvements musculaires.
Les mutations dans ces gènes peuvent entraîner une accumulation anormale de protéines, ce qui peut endommager les neurones moteurs et entraîner leur mort progressive. Cette perte de neurones moteurs est responsable des symptômes caractéristiques de la maladie de Charcot, notamment la faiblesse musculaire, la paralysie et la difficulté à parler, à avaler et à respirer.
Quels sont les symptômes de la maladie de Charcot
Au début de la maladie, les symptômes de la sclérose latérale amyotrophique (SLA) peuvent être indolores. Cependant, ils progressent lentement pour causer une faiblesse musculaire et une atrophie associée à des fasciculations, qui sont des secousses musculaires spontanées. Les patients atteints de SLA peuvent souvent éprouver des crampes et une perte de poids.
Les symptômes de la maladie de Charcot peuvent varier d’une personne à l’autre, mais incluent généralement :
- Faiblesse musculaire,
- Fourmillements,
- Engourdissement des membres,
- Difficultés à parler, avaler et respirer,
- Crampes et spasmes musculaires,
- Perte de poids involontaire.
Qu’est ce qui favorise la SLA ?
Pour le moment, les chercheurs n’ont pas encore réussi à comprendre pourquoi certaines personnes sont touchées par cette maladie tandis que d’autres ne le sont pas. On sait que seulement 10% des cas sont d’origine familiale. « Dans ces situations, une cause génétique est probable, bien qu’elle ne soit pas toujours simple à prouver« , précise le site de l’Inserm.
En ce qui concerne l’environnement des patients, plusieurs pistes sont étudiées, telles que le tabagisme, l’exposition à des pesticides ou à des métaux lourds. Cependant, il n’existe pas encore de données scientifiques confirmant ces hypothèses.
La pratique intense d’un sport de haut niveau pourrait également représenter un facteur de risque. Plusieurs cas ont été signalés chez d’anciens athlètes, comme Jérôme Golmard, un ancien joueur de tennis décédé en 2014. Dans les pays anglophones, la maladie est d’ailleurs appelée « maladie de Lou-Gehrig », en référence à un célèbre joueur de baseball américain décédé de la maladie en 1939. Toutefois, aucune preuve ne permet encore de valider cette hypothèse.
Quel est le diagnostic et l’évolution de la maladie ?
Le diagnostic de la maladie de Charcot repose sur un examen clinique approfondi, des tests électromyographiques et des analyses sanguines pour éliminer d’autres affections. Malheureusement, la SLA est une maladie évolutive et incurable, avec une espérance de vie moyenne de 3 à 5 ans après le diagnostic.
Quelle est l’espérance de vie de la maladie de Charcot ?
L’espérance de vie pour une personne atteinte de la maladie de Charcot (SLA) varie considérablement et dépend de nombreux facteurs, notamment de l’âge de début de la maladie, de la vitesse de progression de la maladie et de la réponse du patient aux soins de soutien.
En général, les personnes atteintes de SLA ont une espérance de vie moyenne de deux à cinq ans à compter de l’apparition des premiers symptômes, bien que certaines personnes puissent vivre plus longtemps. Dans de rares cas, des personnes atteintes de SLA ont vécu jusqu’à dix ans ou plus après le diagnostic.
Il est important de noter que chaque personne atteinte de SLA a une expérience différente de la maladie, et il est difficile de prédire avec précision la durée de vie d’un patient individuel. Le traitement précoce des symptômes, la gestion de la nutrition et de la respiration, ainsi que le soutien émotionnel et social peuvent aider à améliorer la qualité de vie et à prolonger la durée de vie des patients atteints de SLA.
Maladie de Charcot : 5 cas dans la même rue d’un village de la Somme
Peut-on guérir de la SLA ?
À ce jour, il n’existe pas de traitement curatif connu pour la maladie de Charcot. Toutefois, un traitement médicamenteux appelé riluzole peut ralentir légèrement ou modérément la progression de la maladie, ce qui peut retarder le besoin d’une assistance respiratoire chez les patients. Le riluzole agit en ralentissant la mort des motoneurones, bien que le processus exact ne soit pas complètement compris.
Lorsque la perte de la capacité respiratoire devient significative, une assistance respiratoire est nécessaire, d’abord avec un masque pour la respiration non invasive, puis avec une trachéostomie. De plus, une prise en charge pluridisciplinaire et personnalisée est nécessaire, comprenant des séances de kinésithérapie pour stimuler les muscles, des séances d’orthophonie pour améliorer la parole et la déglutition, et de l’ergothérapie pour adapter l’environnement du patient et favoriser son autonomie, ainsi qu’une modification de l’alimentation pour faciliter la déglutition.
Les recherches se poursuivent pour développer un traitement efficace pour la maladie de Charcot. Différentes pistes sont explorées, telles que l’utilisation d’un antiépileptique appelé ezogabine, des thérapies antisens pour réduire le taux de SOD1 muté, et des recherches en thérapie cellulaire.
Avancées thérapeutiques pour la maladie de Charcot
Bien qu’il n’existe pas encore de traitement curatif pour la maladie de Charcot, plusieurs avancées thérapeutiques ont été réalisées pour ralentir la progression de la maladie et améliorer la qualité de vie des patients. Parmi elles :
- Riluzole : un médicament approuvé pour ralentir la progression de la SLA en réduisant la libération de glutamate, un neurotransmetteur qui peut endommager les neurones moteurs.
- Édavarone : un autre médicament approuvé pour la SLA qui agit en neutralisant les radicaux libres responsables de l’endommagement des cellules.
- QALSODY™ (tofersen) : un traitement récemment autorisé par la FDA pour les patients atteints de la maladie de Charcot ayant une mutation du gène SOD1, avec des résultats prometteurs.
QALSODY™ le traitement qui cible les causes génétiques de la SLA
QALSODY™ est le tout premier médicament approuvé aux États-Unis pour traiter une cause génétique de la SLA. Biogen a travaillé avec Ionis Pharmaceuticals pour développer ce traitement, appelé tofersen.
Des effets secondaires graves
Cependant, il est important de noter que QALSODY™ peut entraîner des effets secondaires graves, tels que des problèmes neurologiques, une inflammation de la moelle épinière, un gonflement du nerf optique, une pression accrue dans le crâne et une méningite non infectieuse. Si des symptômes liés à ces problèmes apparaissent, il est crucial d’effectuer un diagnostic et un traitement appropriés. Dans certains cas, il peut être nécessaire d’arrêter ou de suspendre temporairement le traitement par QALSODY™.
Parmi les effets indésirables courants, on retrouve des douleurs, une fatigue extrême, des douleurs articulaires, un taux élevé de globules blancs dans le liquide céphalorachidien et des douleurs musculaires. Ces effets secondaires ont été observés chez 10% ou plus des patients sous QALSODY™ et plus fréquemment que chez ceux recevant un placebo.
« Depuis que les mutations du gène SOD1 ont été identifiées comme une des causes de SLA pour la première fois il y a 30 ans, les spécialistes de la SLA de forme familiale sont en quête de traitements ciblant les facteurs génétiques. QALSODY™ est un traitement ciblant la cause sous-jacente de la SLA-SOD1 qui frappe des familles touchées par cette maladie dévastatrice depuis des générations et qui ont perdu des parents dans la fleur de l’âge. Nous vivons aujourd’hui un moment marquant dans la recherche sur la SLA car QALSODY™ est le premier traitement de la SLA approuvé sur la base d’un biomarqueur », affirme Jean Swidler, président de Genetic ALS & FTD : End the Legacy. « Nous sommes impatients de voir quelles thérapies futures seront développées maintenant que la diminution de la concentration en neurofilaments est admise comme preuve importante de l’effet du traitement sur le processus neurodégénératif. »
Quelle est l’efficacité du QALSODY™ ?
 L’efficacité du médicament QALSODY™ a été testée lors d’une étude clinique sur des patients âgés de 23 à 78 ans souffrant de faiblesse musculaire liée à la SLA et présentant une mutation spécifique du gène SOD1. Au total, 108 patients ont participé à l’étude, qui a duré 24 semaines, et ils ont été divisés en deux groupes : l’un recevant QALSODY™ et l’autre un placebo.
L’efficacité du médicament QALSODY™ a été testée lors d’une étude clinique sur des patients âgés de 23 à 78 ans souffrant de faiblesse musculaire liée à la SLA et présentant une mutation spécifique du gène SOD1. Au total, 108 patients ont participé à l’étude, qui a duré 24 semaines, et ils ont été divisés en deux groupes : l’un recevant QALSODY™ et l’autre un placebo.
Au cours de l’essai, les patients traités par QALSODY™ ont montré une progression moins importante de leur maladie par rapport au groupe placebo, sans toutefois obtenir de résultats statistiquement significatifs. Cependant, QALSODY™ a entraîné une réduction significative de certaines substances dans le sang et le liquide cérébrospinal des patients, ce qui suggère un effet bénéfique du médicament.
Après 52 semaines, une analyse intermédiaire a été réalisée sur les patients qui ont terminé l’essai et sont passés à une étude d’extension en ouvert. Les résultats ont montré que les patients qui ont commencé à prendre QALSODY™ plus tôt ont eu une baisse plus importante de la progression de la maladie, sans toutefois démontrer une différence statistiquement significative.
Le médicament QALSODY™ a été autorisé sur la base des résultats combinés de ces deux études, publiés dans la revue médicale The New England Journal of Medicine.
« J’ai été témoin des bénéfices de QALSODY™ sur le ralentissement de la progression de la SLA chez les patients présentant une mutation du gène SOD1 » a déclaré Timothy M. Miller, MD, Ph.D, investigateur principal des essais cliniques sur le tofersen et co-directeur du centre SLA de la faculté de médecine de Washington University à Saint Louis, aux États-Unis.
«L’autorisation de QALSODY™ octroyée par la FDA me donne l’espoir que les personnes atteintes par cette forme rare de SLA puissent voir le déclin de leur force, de leur fonction physique et de leur fonction respiratoire ralentir. »
Le nouveau médicament QALSODY sera envoyé aux professionnels de la santé aux États-Unis dans environ une semaine. Biogen prévoit que l’accès à ce traitement variera d’un établissement médical à l’autre, le temps que les médecins et les soignants s’habituent au produit.
Un podcast sur la Maladie de Charcot
En vue de faciliter le diagnostic, les filières de santé maladies rares et les réseaux européens mettent leur expertise à disposition pour informer tant les patients que les professionnels de santé. C’est également le but que RARE à l’écoute s’est assigné. Le podcast, à travers une série de cinq épisodes sur la maladie de Charcot, convie des professionnels de santé ainsi que l’association de patients ARSLA pour partager leurs expériences. Leurs précieuses connaissances permettent un échange ouvert et sensibilisent à la fois les praticiens et le grand public sur cette maladie qui reste largement méconnue. Le premier épisode, qui inclut une intervention du Professeur Philippe Couratier, sera disponible à partir du 25 mai sur les principales plateformes de podcast, y compris Apple Podcasts, Spotify, Deezer, Google Podcasts et Amazon Music.
Sources :
- QALSODY Prescribing Information, Cambridge, MA:
- National Institute of Neurological Disorders and Stroke. Amyotrophic Lateral Sclerosis (ALS). Available at: https://www.nindnih.gov/health-information/disorders/amyotrophic-lateral- sclerosis-als. Accessed: April 2023.
- Frederiksen SD, Avramović V, Maroilley T, et al. Rare disorders have many faces: in silico characterization of rare disorder spectrum. Orphanet J Rare Dis. 2022 Feb 22;17(1):76. doi: 1186/s13023-022-02217-9.
- Hee SW, Willis A, Tudur Smith C, et al. Does the low prevalence affect the sample size of interventional clinical trials of rare diseases? An analysis of data from the aggregate analysis of gov. Orphanet J Rare Dis. 2017 Mar 2;12(1):44. doi: 10.1186/s13023-017-0597-1.
- Brown CA, Lally C, Kupelian V, Flanders Estimated Prevalence and Incidence of Amyotrophic Lateral Sclerosis and SOD1 and C9orf72 Genetic Variants. Neuroepidemiology. 2021;55(5):342- 353. doi: 10.1159/000516752. Epub 2021 Jul 9.
- Yuan A, Rao MV, Veeranna, Nixon Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb Perspect Biol. 2017;9(4):a018309.
- Miller TM, Cudkowicz ME, Genge A, et Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2022;387:1099-110. doi: 10.1056/NEJMoa2204705.
- Akcimen F, Lopez ER, Landers JE, et Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat Rev Genet. 2023. https://doi.org/10.1038/s41576-023-00592-y
Sophie Madoun


{kind=link}