L’hémochromatose génétique (HG) est due à une mutation du gène HFE qui provoque une accumulation de fer dans l’organisme. Avec le temps, le fer en surcharge altère le foie, le pancréas, le cœur, les glandes endocrines, les articulations. Première maladie génétique, elle atteint 1 Français sur 300. Mais l’expression de la maladie est très variable. Certains patients vont avoir une maladie atténuée, d’autres très grave pouvant entraîner cirrhose, cancer, insuffisance cardiaque, diabète… Un diagnostic précoce, à 20-35 ans, permet d’éviter ces graves manifestations.
MÉCANISME DE L’HÉMOCHROMATOSE GENETIQUE
1) Chez un sujet normal
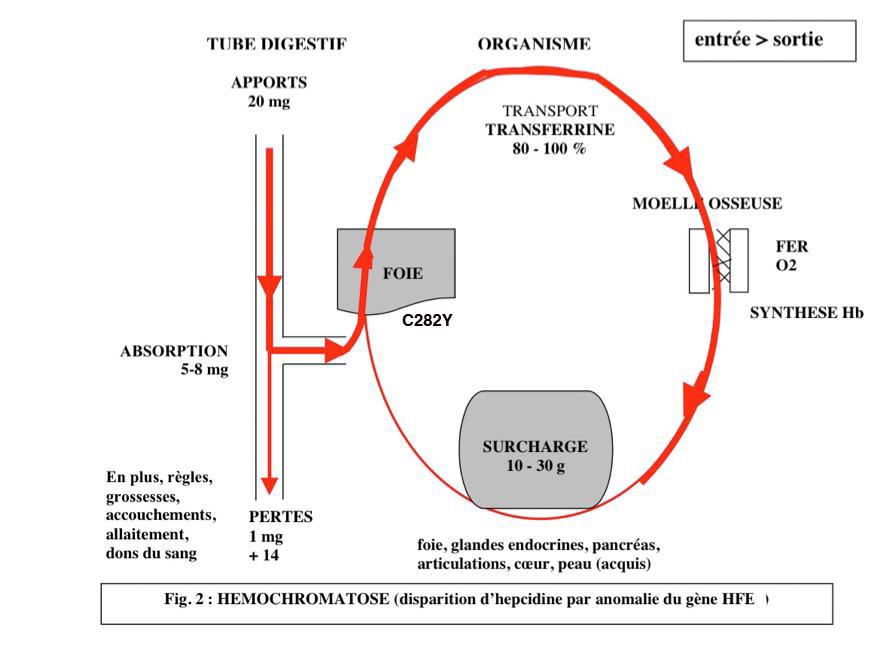
tous les jours 1 mg de fer est absorbé au niveau du tube digestif sur les 20 mg apportés par l’alimentation. Ce fer est transporté vers la moelle osseuse grâce à une protéine : la transferrine, normalement saturée à 30 %. Il entre dans la composition de l’hémoglobine des globules rouges. Sur ce fer se fixe l’oxygène des poumons. Les globules rouges ont une durée de vie de 120 jours ; le fer alors libéré est récupéré et sert à la synthèse de nouveaux globules rouges. Il est donc réutilisé en permanence. Seul 1 mg est perdu tous les jours par voie urinaire, digestive, et par la peau. Donc 1 mg est absorbé, 1 mg est éliminé. (fig. 1)

2) Chez le sujet atteint de la maladie
l’absorption du fer est de 5 à 6 mg par jour par absence d’hepcidine (conséquence de la mutation génétique), et 1 mg seulement est perdu (sauf chez la femme : règles, grossesses, allaitement). La transferrine est dite saturée à 60-80-100 %. Les réserves sont débordées, le fer se dépose dans les organes tous les jours un peu plus, le foie, le pancréas, le cœur, les articulations… où il provoque altérations fonctionnelles et anatomiques (par exemple, destruction des îlots de Langerhans du pancréas par les radicaux libres créant un diabète par absence de sécrétion d’insuline). (fig. 2)
LE GÈNE HFE RESPONSABLE DE L’HÉMOCHROMATOSE
Il a été découvert en 1996. Il certifie l’hémochromatose génétique. Il est fixé sur le chromosome 6. Les mutations du gène HFE sont au nombre de deux. Elles provoquent l’absence d’hepcidine. Il s’agit de la mutation C282Y et de la mutation H63D. Ces mutations peuvent être présentes à l’état homozygote C282Y++/C282Y++ ou hétérozygote composite C282Y+-/H63D+-. Les mutations hétérozygotes C282Y ou H63D, isolées, ne donnent pas la maladie.
LE DIAGNOSTIC DE L’HÉMOCHROMATOSE GÉNÉTIQUE
C’est la difficulté principale. Cette maladie est le plus souvent méconnue à 20-35 ans, âge où il faudrait faire le diagnostic pour conserver une espérance de vie normale. Elle est souvent diagnostiquée à 50-70 ans suite à des complications qui égarent le malade en diabétologie, cardiologie, hépatologie…
1) Le diagnostic doit être fait à 20-35 ans
À cet âge, il y a peu de fer en surcharge en fer (5 à 8 g). Cliniquement, les symptômes ne sont pas évidents mais il est cependant « anormal qu’un patient de cet âge » se plaigne de :
fatigue intense, épuisement obligeant à l’arrêt de l’activité et pouvant entraîner un état dépressif ;
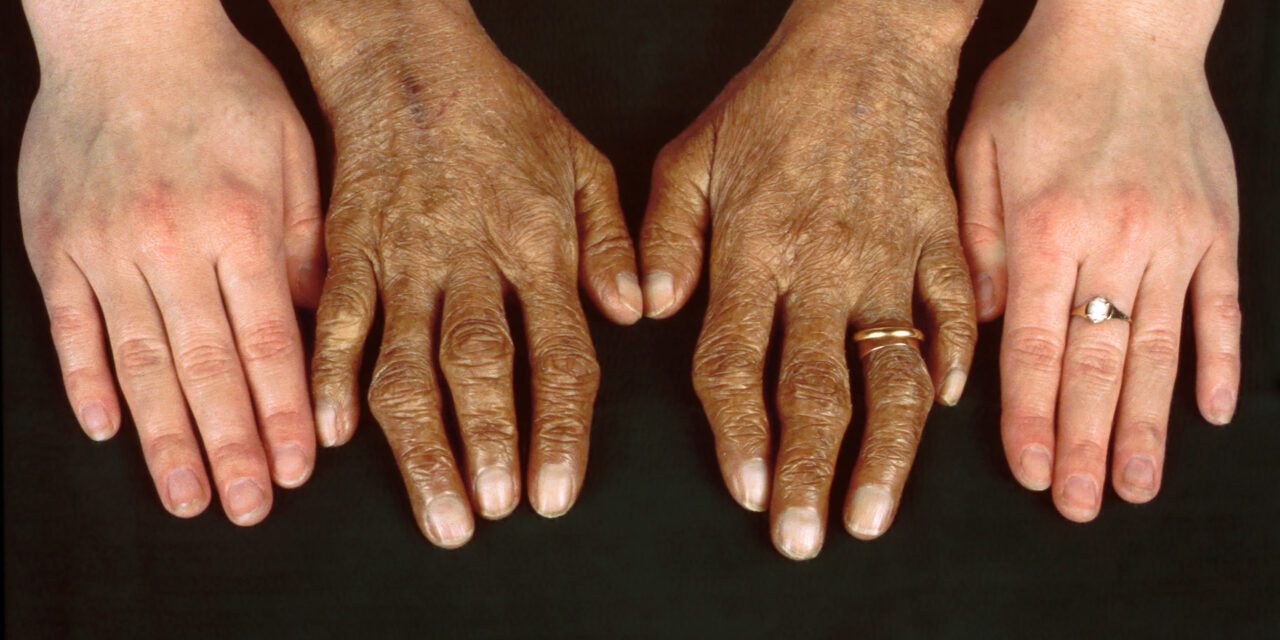
douleurs articulaires des mains (articulations métacarpo-phalangiennes des 2ème et 3ème doigts, poignée de mains douloureuse) ou douleurs de chevilles, genoux… ;
troubles sexuels (impuissance, perte de la libido, ménopause précoce) ;
troubles du rythme cardiaque intermittents ;
essoufflement pour un effort minime ;
augmentation intermittente des transaminases et de la glycémie.
Il faut faire une prise de sang pour rechercher ces perturbations biologiques :
1) La saturation de la transferrine (normale jusqu’à 50 %).
2) La ferritinémie (normale de 30 à 300 µg/l).
3) Si ces deux résultats sont anormaux, la mutation C282Y homozygote confirme le diagnostic d’HG.
Le diagnostic étant fait, il faut faire disparaître la surcharge en fer par des saignées :
– Commencer les saignées dont le rythme et le volume sont déterminés en fonction du taux de ferritine.
– Faire un examen du cœur (ECG), du foie (échographie), une radiographie des articulations des mains, des chevilles, doser la glycémie, la TSH, la testostérone.
2) Si le diagnostic est fait à 50-75 ans
la surcharge en fer peut être de 30 à 40 g. En général, les symptômes cliniques égarent le patient chez différents spécialistes qui ne soupçonnent pas toujours l’HG. Ainsi :
– L’asthénie (85 %) domine toujours, elle est permanente, obligeant très vite au mi-temps, à l’invalidité. Elle conduit à un état dépressif. Elle est physique, psychique et sexuelle.
– Les lésions articulaires (75 %) sont douloureuses, multiples, atteignant petites et grandes articulations. Il y a parfois des phénomènes inflammatoires, de véritables accès goutteux, des pincements articulaires, des géodes osseuses, une condensation sous-chondrale avec lésion de chondrocalcinose. L’ostéoporose est fréquente. Les prothèses articulaires sont souvent indispensables (hanche, genou, épaule).
– L’atteinte hépatique (20 %) se traduit par une augmentation du volume du foie témoignant de la surcharge en fer. Les tests hépatiques sont peu perturbés mais la cirrhose est toujours envisagée lorsque la ferritine est supérieure à 1 000 µg/l. Une échographie du foie et un dosage de l’alpha-foetoprotéine doivent être faits tous les 6 mois dans ce cas pour surveiller l’apparition d’un cancer du foie.
– Le diabète sucré (15 %) est insulino-dépendant, du à la destruction des îlots de Langerhans. L’insulino-résistance due à la cirrhose aggrave encore ce diabète.
– L’atteinte cardiaque (15 %) est grave par ses troubles du rythme (fibrillation, flutter) et une insuffisance cardiaque.
– Les lésions des glandes endocrines (hypophyse, thyroïde, glandes sexuelles) expliquent l’asthénie et les troubles sexuels.
– Les signes cutanés évidents sont la mélanodermie avec le temps, plus rare les anomalies des ongles, une dépilation, des cheveux secs et cassants.
Toutes ces « complications », dues au retard de diagnostic, font de l’HG une maladie grave et même mortelle (2 000 décès par an).
LE TRAITEMENT DE L’HÉMOCHROMATOSE GÉNÉTIQUE
Ce sont les saignées, traitement simple, peu onéreux. Le but est à la fois d’éliminer l’excès de fer (phase d’attaque) et d’éviter la reconstitution de la surcharge (phase d’entretien). Ce traitement dure en général toute la vie sauf chez 10 % des patients qui n’ont plus besoin de saignées après plusieurs années.
Il est recommandé de faire les 5 premières saignées dans une structure de soins ; les saignées à domicile sont ensuite possibles. Le volume prélevé doit s’adapter à la tolérance du patient, à son âge, à son état de santé (insuffisance cardiovasculaire)… À noter, depuis 2009, dans les EFS disposant d’un centre de santé, le sang peut être conservé et non plus détruit ; la saignée devient un « don-saignée ».
Il est recommandé la rédaction d’un projet thérapeutique. L’infirmière doit être présente en permanence et un médecin doit être disponible rapidement. La pression artérielle et la fréquence cardiaque doivent être prises avant et après la saignée, le patient être en position semi-allongée, la saignée doit prendre 15 mn ; la ponction se fait à partir d’une veine du pli du coude. Le patient doit rester en position déclive pendant ½ heure, recevoir un apport liquide et/ou alimentaire avant de partir. Tout incident (malaise, perte de connaissance) doit être signalé à son médecin. Les saignées peuvent être réalisées chaque semaine ou tous les 15 jours ; elles sont espacées dans le temps au fur et à mesure que la ferritine baisse. On doit atteindre une ferritine entre 50 et 100 µg/l à la fin de la phase d’attaque avec une hémoglobine supérieure à 11 g/dl. Au-dessous de ce chiffre les saignées sont arrêtées. La saturation de la transferrine ne sert qu’à faire le diagnostic et n’intervient pas dans la fréquence des saignées.
Il n’y a pas de régime alimentaire, ni d’autres médicaments. Le thé est recommandé sans être démontré efficace.
INTÉRÊT DU DÉPISTAGE FAMILIAL
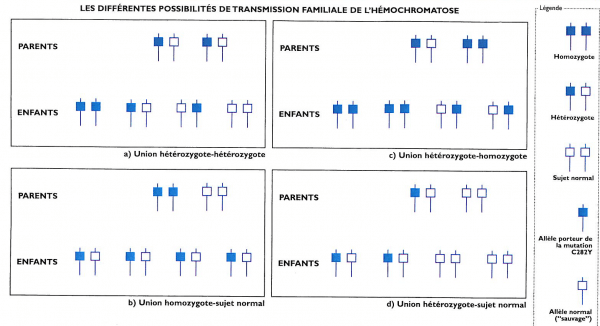
L’hémochromatose génétique est une maladie héréditaire récessive (transmission non obligatoire). Le patient atteint ou « probant » doit signer un consentement pour le test génétique demandé par le médecin. Le médecin prescripteur reçoit le résultat et a la charge de l’expliquer à son patient mais ne peut pas le dire à la famille. Seul le probant peut avertir les membres de sa famille qui doivent contrôler saturation de la transferrine et ferritine. (tableau 1).
CONCLUSION
L’hémochromatose génétique est une maladie encore méconnue dont le diagnostic est fait trop tardivement et qui reste grave de ce fait. C’est un problème de santé publique non résolu. Il y a quand même 2 000 morts par an qui pourraient être évitées si un dépistage systématique était fait. C’est le seul moyen de voir disparaitre cette maladie. Ainsi un diagnostic précoce à 20-35 ans, suivi de saignées, permet d’avoir une espérance de vie normale. Il faut faire au moins une fois dans sa vie (au cours de consultation, par exemple), un dosage du coefficient de saturation de la transferrine et la ferritine.
POUR EN SAVOIR PLUS :
– 1- Hémochromatose génétique : Presse Médicale M. Bismuth, P. Martinez et H. Michel 2003
– 2- Prise en charge de l’hémochromatose liée au gène HFE : HAS recommandations 2005
– 3- European Association for the study of the liver. EASL Clinical Practice Guidelines for HFE hemochromatosis. J. Hepatol (2010)


{kind=link}